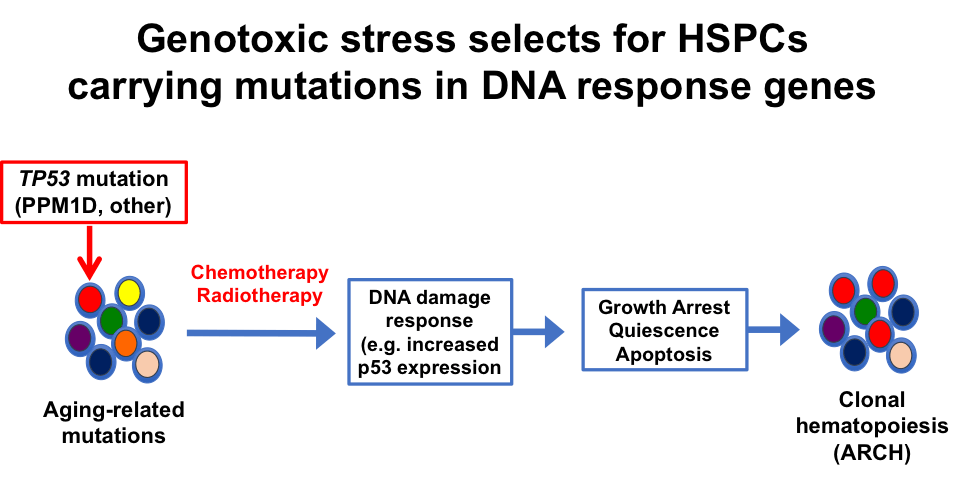
The long-term goal of this project is to identify genetic and epigenetic alterations that contribute to the development of treatment-related acute myeloid leukemia (tAML) and to exploit this knowledge to develop new therapies for this high-risk subset of AML. tAML is a late complication of chemotherapy and/or radiation therapy. Survival is measured in months, not years; thus, there is a pressing unmet clinical need for new therapies for tAML and other therapy-related myeloid neoplasms. Mutations of TP53 are enriched in tAML compared to de novo AML with a frequency of 30-40% and 5-10%, respectively. Mutations of TP53 are associated with a complex karyotype and very poor overall survival, both for de novo and tAML cases. Many tAML cases, however, do not have TP53 mutations; although some have mutations in other AML-initiating genes, many have no well-recognized mutations that contribute to clonal selection by chemotherapy. To date, only 22 tAML genomes have been sequenced. Clearly, additional cases must be characterized to understand the full spectrum of “sensitizing” mutations (like TP53), patterns of cooperating mutations, structural variants, and epigenomic events that contribute to the progression of tAML. Our recent studies show that in a subset of tAML, mutations of TP53 are the initiating mutation. Moreover, we show that certain hematopoietic stressors, such as genotoxic stress from chemotherapy, or ribosome biogenesis stress from inherited mutations in SBDS, result in the selective expansion of hematopoietic stem progenitor cells (HSPCs) carrying heterozygous TP53 mutations. Based on these observations, we hypothesize that both cell-intrinsic events and cell-extrinsic stressors contribute to the development of clonal hematopoiesis. We further hypothesize that repeated genotoxic or hematopoietic stress is key to clonal evolution from clonal hematopoiesis to AML.